数分でゲノム構造を予測:AIが切り拓く新時代のクロマチン解析
特定のDNA配列が細胞核内でどのように配置されるかを予測する新たなアプローチが、これまで数日かかっていた解析をわずか数分で可能にしました。人間の体内のすべての細胞は同じ遺伝情報を持っていますが、実際に発現する遺伝子は細胞ごとに異なります。脳細胞と皮膚細胞が異なる機能を持つのは、三次元的なゲノム構造が各遺伝子のアクセス可能性を調整し、細胞特異的な遺伝子発現パターンを作り出しているためです。

MITの化学者らは、生成型人工知能を用いて、これらの3Dゲノム構造を高速かつ正確に予測する新技術を開発しました。この技術により、従来の実験法よりもはるかに短時間で数千の構造を予測することが可能となり、ゲノムの三次元配置が細胞機能や遺伝子発現にどのような影響を及ぼすのかを解明する研究が加速することが期待されます。
「私たちの目的は、DNA配列から3次元のゲノム構造を予測することでした」と語るのは、MIT化学科の准教授であり、この研究の責任著者であるビン・チャン博士(Bin Zhang, PhD)です。「この技術は最先端の実験技術と同等の精度を持ち、非常に多くの新しい研究機会を切り開くことができます。」
この研究は、MITの大学院生であるグレッグ・シュエッテ(Greg Schuette)氏とズオハン・ラオ(Zhuohan Lao)氏が筆頭著者を務め、2025年1月31日に『Science Advances』誌に掲載されました。論文タイトルは「ChromoGen: Diffusion Model Predicts Single-Cell Chromatin Conformations(ChromoGen:拡散モデルによる単一細胞クロマチン構造の予測)」です。
配列から構造へ:DNAの折りたたみをAIで予測
細胞核内では、DNAとタンパク質が結合してクロマチンと呼ばれる複合体を形成しています。クロマチンは複数の階層構造を持ち、約2メートルのDNAが直径0.01ミリメートルの核内に収まるための仕組みでもあります。DNAはヒストンと呼ばれるタンパク質に巻き付いており、その構造は「糸に通されたビーズ」のような形に例えられます。
このクロマチン構造に影響を与えるのが、エピジェネティック修飾と呼ばれる化学的なタグです。これらの修飾は細胞ごとに異なり、DNAの特定位置に付加されることで、クロマチンの折りたたみ方や周囲の遺伝子のアクセス性に変化を与えます。つまり、クロマチン構造の違いは、どの遺伝子がどの細胞で発現されるかを決定づける要因の一つです。
過去20年にわたり、科学者たちはクロマチン構造を決定するための実験技術を開発してきました。その中でも広く使われているのが、Hi-C法です。この手法では、細胞核内で近接しているDNA断片を化学的に連結し、その後DNAを細かく断片化して配列決定(シーケンシング)を行い、どの部分が隣接していたのかを特定します。
この方法は、複数の細胞を用いて平均的なクロマチン構造を調べたり、単一細胞に特化して個別の構造を明らかにすることもできます。しかし、Hi-Cや関連手法は労力と時間がかかり、1細胞のデータを得るのに1週間程度かかるのが一般的です。
このような制約を克服するために、チャン博士と学生たちは、生成AIの進歩を活用し、単一細胞におけるクロマチン構造を高速かつ正確に予測するモデルを開発しました。このAIモデルはDNA配列を解析し、そこから細胞内で形成される可能性のあるクロマチン構造を予測することができます。
「ディープラーニングはパターン認識に非常に優れているのです」とチャン博士(Bin Zhang PhD)は語ります。「この技術を使えば、数千塩基対にわたる非常に長いDNA配列を解析し、それらの塩基対にどんな重要な情報がコードされているのかを見つけ出すことができます。」
研究者らが開発したモデル「ChromoGen」には、2つの主要な構成要素があります。
1つ目の構成要素は深層学習モデルで、ゲノムを「読む」ように設計されています。このモデルは、DNA配列に含まれる情報と、細胞型に特異的なクロマチンアクセシビリティデータ(広く利用可能な公開情報)を解析します。
2つ目は、生成AIモデルで、物理的に妥当なクロマチン構造を予測します。このモデルは、1,100万以上のクロマチン構造データを用いて学習されました。これらのデータは、Dip-C法(Hi-C法の一種)を用いて、人のBリンパ球系列由来の16細胞から取得されたものです。
この2つの構成要素が統合されることで、細胞型に特異的な環境がクロマチン構造の形成にどう影響するかを生成モデルに伝えることができ、DNA配列と立体構造の関係性を効果的に捉えることが可能になります。研究者らは各配列ごとに、多数の構造をモデルから生成しています。これは、DNAが非常に無秩序な分子であるため、1つの配列から多様な構造が生じる可能性があるためです。
「ゲノム構造の予測を難しくしている大きな要因は、正解が1つではないという点です。どのゲノム領域においても、構造には分布が存在します。この高次元で複雑な統計的分布を予測するのは、非常に難しい課題なのです。」とシュエッテ氏(Greg Schuette)は語ります。
高速な解析を実現
一度学習を終えたモデルは、Hi-C法や他の実験手法よりもはるかに速い時間スケールで予測を生成できます。
「従来であれば、ある細胞型について数十個の構造を得るために6か月の実験が必要でしたが、私たちのモデルなら、GPU 1台で20分以内に特定領域の構造を1000個生成できます。」とシュエッテ氏は述べています。
モデルの学習後、研究者らは2000以上のDNA配列に対して構造予測を行い、それを実験的に得られた構造と比較しました。その結果、AIモデルによって生成された構造は、実験データと一致、もしくは非常に類似していることが判明しました。
「私たちは通常、1つの配列に対して数百から数千の構造を観察します。これにより、その領域で形成されうる構造の多様性を十分に表現することができます」とチャン博士は語ります。「同じ実験を異なる細胞で何度も行えば、おそらく非常に異なる構造に出会うでしょう。それこそが、私たちのモデルが予測しようとしているものなのです。」
さらにこのモデルは、学習に使用していない細胞型のデータに対しても高い予測精度を示しました。これは、細胞型ごとのクロマチン構造の違いや、それが機能に与える影響の解明に役立つ可能性があります。また、1つの細胞内に存在する異なるクロマチン状態の探索や、それが遺伝子発現に与える変化の解析にも利用できます。
さらに、特定のDNA配列に生じた突然変異がクロマチン構造にどのような変化をもたらすかを調べることも可能であり、疾患の発症メカニズムの理解にもつながると期待されています。
「このようなモデルを使えば、非常に多くの興味深い疑問に取り組むことができると思います。」とチャン博士は語ります。
研究チームは、本モデルおよびすべてのデータを、利用を希望する他の研究者にも公開しています。
本記事は、MITのアン・トラフトン氏(Anne Trafton)によるプレスリリースをもとに構成しています。
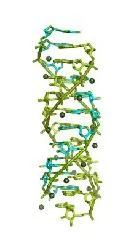
画像:この画像はDip-C研究で報告されたいくつかの染色体の3次元ゲノム構造を示しており、新しいChromoGenモデルのトレーニングに使用された。(Credit: Courtesy of the researchers; edited by MIT News)
[MIT news release] [Science Advances article]