2025年1月16日付で『Cell』誌に掲載された新たな研究論文「Long Somatic DNA-Repeat Expansion Drives Neurodegeneration in Huntington’s Disease(長い体細胞DNAリピートの拡大がハンチントン病の神経変性を引き起こす)」では、ハンチントン病(HD)について詳細に検討されており、その進行がHTT遺伝子内のCAGリピートの体細胞内拡大と特定の神経細胞において関連していることが明らかになりました。この研究は、ハーバード・メディカル・スクールおよびマサチューセッツ工科大学・ハーバード大学のブロード研究所に所属するロバート・E・ハンズエイカー(Robert E. Handsaker)、セヴァ・カシン(Seva Kashin)、サビーナ・ベレッタ(Sabina Berretta)、スティーブン・A・マッキャロール(Steven A. McCarroll)ら研究者によって主導されました。本研究は、HDの特徴的な神経細胞変性、特に線条体投射ニューロン(SPN)におけるメカニズムに光を当てるものです。この成果は、HDに関する重要な疑問を明らかにするとともに、同様の遺伝性疾患に対する新たな治療方針を示唆しています。









ハンチントン病のメカニズム
HDは、HTT遺伝子内のCAGリピート配列の生殖細胞系列における拡大によって引き起こされます。通常の人ではこのリピートは15〜30回ですが、HDを発症する人では36回以上となっています。この拡大はハンチンチン(huntingtin)タンパク質内のポリグルタミン配列をコードし、これが疾患の発症に関与していると考えられています。しかしながら、遺伝的に受け継がれたリピートの長さが発症リスクや発症年齢を決定する一方で、どのようにして神経細胞死が起こるのかは長年の謎でした。
本研究では、「体細胞内リピート拡大」(somatic repeat expansion)―つまり、受け継がれたCAGリピートが時間とともにさらに増加する現象―が神経変性の主な要因であることが示されました。研究者らはシングルセル解析を用いて、以下の重要な事実を明らかにしました。
SPNが特異的に影響を受ける:
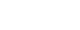
線条体投射ニューロン(striatal projection neurons:SPNs)は、線条体の主要な神経細胞であり、最も顕著な体細胞内リピート拡大が観察されました。
HDの影響を受けた個体では、SPNにおけるCAGリピートは、遺伝的に受け継がれた長さ(多くの場合40〜45リピート)を超え、個々のニューロンでは最大で800回にまで達していました。
有害な閾値が存在する:
CAGリピートが150回を超えるニューロンでは、ポジティブな神経細胞マーカーの発現が失われ、細胞老化およびアポトーシス経路が活性化され、最終的に変性が進行します。
一方で、この閾値未満(150リピート以下)の拡大を持つニューロンは、かなりの拡大があっても、遺伝子発現のプロファイルはごくわずかな拡大を持つニューロンと類似していました。
細胞型特異性と選択性:
SPNは体細胞リピート不安定性に対して特に感受性が高い一方で、線条体内の他の細胞型(介在ニューロンやグリア細胞など)は拡大がわずかであり、変性を免れていました。
この細胞型特異性こそが、HDにおける線条体の選択的病理を説明しています。
ゆっくりと進むタイマー
HDの進行には、体細胞内のCAGリピートが徐々に拡大していく長い潜伏期間が関与しています。このプロセスは以下のような段階に分かれます:
フェーズA(40〜80 CAG):拡大は非常に緩やかに進み、数十年かけて中程度の長さに達します。
フェーズB(80〜150 CAG):拡大がより速く進行し、有害な閾値に近づきます。
フェーズC(150〜250 CAG):150回を超えると、ニューロンは劇的な変化を示し、細胞のアイデンティティを喪失し、数百の遺伝子の調節異常が起きます。
フェーズD(250 CAG超):ニューロンは危機段階に入り、SPNでは通常抑制されている発生関連遺伝子やアポトーシス促進遺伝子(例:CDKN2AおよびCDKN2B)を活性化させます。
フェーズE(神経細胞死):最終的にニューロンは変性し、HDに特徴的なSPNの喪失が生じます。
研究者らは、これらの段階によってHDの数十年にわたる無症候性期間が説明できると述べています。多くのニューロンは初期段階にとどまり、有害な拡大に達するまでに長い時間がかかるためです。
研究手法と主要な観察結果
これらの発見を明らかにするために、研究チームは革新的なシングルセル解析技術を用い、個々のニューロンにおけるCAGリピートの長さとRNA発現プロファイルの両方を測定しました。主要な観察結果は以下のとおりです:
拡大は非同期に発生:
ある時点で見た場合でも、大多数のニューロンは依然として無害なHTT(150リピート未満)を保持している一方で、ごく一部のニューロンが有害なリピート長に達していました。
SPNはアイデンティティを喪失:
CAGリピートが150回を超えた拡大を示すニューロンでは、SPN特異的な遺伝子発現が失われました。これには、カリウムチャネルサブユニット(KCNA4, KCND2)やカルシウムシグナル関連遺伝子(RYR3)などが含まれます。
危機遺伝子の活性化:
進行の後期段階(フェーズD)にあるニューロンでは、かつて抑制されていた遺伝子群が活性化されました。その中には、HOXAおよびHOXCクラスター遺伝子など、発生初期に関わるものが含まれています。
治療への示唆
この研究は、ハンチントン病に対する治療戦略の見直しを提案しています。現在多くの治療法は、ハンチンチンタンパク質の量を低下させることを目的としていますが、これはこのタンパク質自体が本質的に毒性を持つという仮説に基づいています。
しかしながら、この研究は、ほとんどのニューロンがCAGリピート数150回に達するまで毒性のあるハンチンチンを産生していないことを示唆しています。これは、別の介入ポイントの存在を示しています。
リピート長の安定化:
体細胞内のリピート拡大を遅らせることができれば、毒性発現のタイミングを遅延させる可能性があります。
たとえ拡大速度をわずかに減少させるだけでも、無症候性の期間を数年延長することができるため、非常に大きな治療上の利益が期待されます。
介入のタイミング:
HDの初期段階では、ほとんどのニューロンが依然として非毒性なHTTアレルを保持しています。
この段階でDNAメンテナンス経路(例えばミスマッチ修復:mismatch repair)を標的にすることで、症状発現後であっても疾患進行を停止あるいは遅延させることができる可能性があります。
より広い疾患への関連:
体細胞内拡大のダイナミクスに関する今回の知見は、他のリピート拡大型疾患にも応用できる可能性があります。
たとえば、筋強直性ジストロフィー、脊髄小脳失調症、フリードライヒ運動失調症など、多くが同様の遅発性かつ進行性の経過をたどります。
未解明だった疑問の解消
この研究は、ハンチントン病に関する以下の長年の謎を解決しました:
なぜ疾患は細胞型特異的なのか?
SPNが体細胞リピート拡大を起こしやすく、他の細胞型はそうではないためです。
なぜ発症までに数十年かかるのか?
有害な150リピートに達するまでには、ニューロンが数十年にわたり拡大を蓄積する必要があるためです。
HD原因アレルはなぜ有害なのか?
遺伝的に受け継がれたアレル自体には有害性がなく、体細胞内の拡大が臨界長を超えることで初めて毒性が発現するためです。
結論
この研究は、ハンチントン病をDNA駆動型のプロセスとして再定義し、体細胞内のCAGリピート拡大こそが神経変性のタイミングと程度を決定することを明確に示しました。
拡大メカニズムに焦点を当てることで、研究者らはHDおよび関連疾患の治療戦略を根本的に変える可能性のある新たな介入機会を特定しました。
この深い理解を基に、次世代の治療法は、単に症状を治療するだけでなく、神経変性の発症そのものを防ぐことが可能になるかもしれません。