物理ベースの機械学習が拓く、新たな生体分子設計の可能性
AIによるタンパク質設計は近年急速に進歩していますが、ノーベル賞を受賞したAlphaFoldのような最先端のAIでも予測が困難な領域が残されています。ヒトのタンパク質の約30%を占める「天然変性タンパク質」は、特定の形を持たずに機能する不思議な存在です。この未開拓領域に対し、ハーバード大学とノースウェスタン大学の研究チームが、物理学に基づいた新しい機械学習の手法を開発しました。この技術は、これまで設計が難しかった生体分子に新たな可能性をもたらすかもしれません。
合成生物学や構造生物学において、人工知能の進歩は、コンピューターを用いて任意のアミノ酸配列の3D構造を正確に予測することにより、抗体から血液凝固因子に至るまで、特定の機能を持つ新しいタンパク質設計の爆発的な進展をもたらしました。
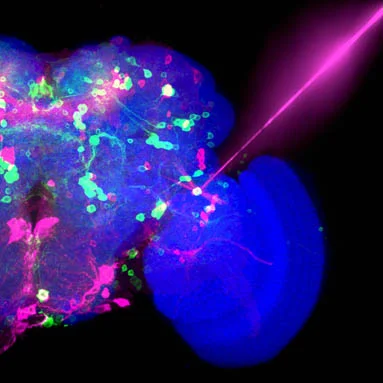
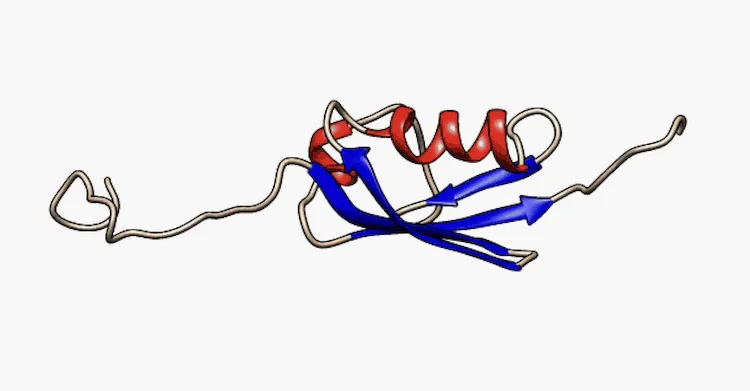
しかし、ヒトゲノムによって発現する全タンパク質の30%近くは、AlphaFoldを含む最も強力なAIツールでさえも、その構造予測が困難です。これらの、いわゆる天然変性タンパク質(IDPs: intrinsically disordered proteins)は、決して固定された形状に収まらず、絶えず動き回っています。これらは分子の架橋、センシング、シグナル伝達といった無数の生物学的機能に不可欠ですが、その固有の不安定さゆえにゼロから設計することが難しいとされてきました。
今回、ハーバード大学ジョン・A・ポールソン工学・応用科学スクール(SEAS)とノースウェスタン大学のチームが、特定の特性に合わせて天然変性タンパク質を設計できる新しい機械学習手法を実証しました。この研究は、これらの謎に満ちた生体分子への新たな理解と、病気の起源や治療法に関する新しい洞察への扉を開くものです。
この研究成果は、2025年10月6日に『Nature Computational Science』誌に掲載されました。SEASの大学院生であるライアン・クルーガー氏(Ryan Krueger)と、元NSF-Simons QuantBioフェローで現在はノースウェスタン大学の助教であるクリシュナ・シュリニバス氏(Krishna Shrinivas)が共同筆頭著者であり、SEASの応用数学・応用物理学Catalyst Professorであるマイケル・ブレナー博士(Michael Brenner, PhD)との共同研究によるものです。論文タイトルは「Generalized Design of Sequence–Ensemble–Function Relationships for Intrinsically disordered Proteins(天然変性タンパク質の配列・アンサンブル・機能関係の一般化設計)」です。
シュリニバス氏は、天然変性タンパク質(IDPs)が、明確な立体構造を持つタンパク質を予測・設計するためのGoogle DeepMindのAlphaFoldのような現在のAIベースの手法では扱えない領域であるため、その研究に興味を持ったと述べました。にもかかわらず、このような変性タンパク質は生物学の多くの基本的側面に重要であり、これらのタンパク質の変異ががんや神経変性疾患に関連していることが知られています。変性タンパク質の一例として、パーキンソン病やその他の疾患に長年関与が指摘されてきたアルファシヌクレインがあります。合成用途や治療用途のためにIDPsを設計するには、「より優れたAIモデルを考案するか、あるいは、良好な予測が得られるだけでなく、物理法則も自動的に手に入れられるような物理モデルを実際に活用する方法を考案する必要があった」とシュリニバス氏は語ります。
自動微分アルゴリズム
この論文では、「自動微分(automatic differentiation)」、すなわち導関数(瞬時変化率)の自動計算を実行できるアルゴリズムを活用した計算手法について説明しています。これは、望ましい挙動や特性を持つタンパク質配列を合理的に選択するために使用されます。この技術は、ディープラーニングやニューラルネットワークのトレーニングに広く使われているツールですが、ブレナー博士の研究室は、物理ベースの分子動力学シミュレーションの最適化といった、他の潜在的なユースケースをいち早く認識していたグループの一つです。
自動微分を用いることで、研究チームは、タンパク質配列のわずかな変化(単一アミノ酸の変化でさえも)が、タンパク質の最終的に望まれる特性にどのように影響するかをコンピューターが認識できるように訓練することができました。彼らはこの手法を、ある機能(例えば、ループやコネクタを作る、あるいは環境中の異なるものを感知するなど)を実行するために必要な基準に適合するアミノ酸配列を見つけ出す、非常に強力な検索エンジンのようだと例えています。
「私たちは、大量のデータを集めて機械学習モデルを訓練し、タンパク質を設計するという方法は取りたくありませんでした」とクルーガー氏は言います。「私たちは、既存の十分に正確なシミュレーションを活用し、それらのシミュレーションのレベルでタンパク質を設計できるようにしたかったのです。」
この手法は、勾配ベースの最適化と呼ばれるニューラルネットワーク訓練の伝統的なフレームワークを活用し、新しいタンパク質配列を効率的かつ正確に特定します。その結果、研究チームが設計したタンパク質は「微分可能」なものとなりました。つまり、それらはAIによって予測された最良の推測ではなく、タンパク質が自然界で実際にどのように動的に振る舞うかを考慮に入れた、実世界の物理学を用いた分子動力学シミュレーションに基づいているのです。