私たちの脳を巨大な都市に例えるなら、脳細胞(ニューロン)は無数の家、そしてそれらをつなぐシナプスは複雑な通信網です。アルツハイマー病やパーキンソン病といった神経変性疾患では、この通信網に深刻な障害が発生します。脳細胞が死に至るずっと前から、シナプスで発生する「タンパク質のゴミ」を掃除する繊細な仕組みが壊れ始め、通信が滞ってしまうのです。この「ゴミ詰まり」が思考や記憶、言語といった私たちの根幹をなす機能の流れを徐々に蝕んでいきます。今回、このゴミ詰まりを防ぎ、脳の通信網を守るための新しい戦略が、ロックフェラー大学の研究によって明らかにされました。

『PNAS』誌に発表されたこの研究は、「PI31」というタンパク質のレベルを高めることで、神経変性を防ぎ、シナプスの機能を回復させ、さらにはパーキンソン病に似た希少遺伝性疾患を持つハエやマウスの寿命を大幅に延ばせることを示しています。この成果は、アルツハイマー病の治療や、加齢に伴う認知機能の低下を遅らせることにも希望をもたらすかもしれません。この2025年9月16日のオープンアクセス論文は、「「PI31 Expression Is Neuroprotective in a Mouse Model of Early-Onset Parkinsonism(PI31の発現は早期発症型パーキンソニズムのマウスモデルにおいて神経保護作用を持つ)」」と題されています。
「アルツハイマー病やパーキンソン病など、多くの疾患は、少なくとも初期段階においてはシナプス機能不全の病気なのです」と、責任著者であり、ロックフェラー大学アポトーシス・がん生物学研究室の室長であるヘルマン・ステラー(Hermann Steller)博士は言います。「今回、シナプスで不要なタンパク質を除去する方法を示したことで、これが一般的な加齢関連疾患の治療に革命をもたらすことを期待しています。」
アミロイド斑:原因か、それとも症状か?
アルツハイマー病やパーキンソン病の脳に見られるタンパク質の凝集体(プラーク)に、すべての原因を押し付けたくなるのは無理もありません。何十年もの間、この分野は「アミロイド仮説」に支配されてきました。これは、アルツハイマー病の特徴であるベータアミロイド斑やタウタングルといった目に見えるタンパク質の塊が、脳細胞死の直接的な原因であるとする考え方です。しかし、これらのプラークを標的とした治療法が臨床で大きな改善をもたらすことに失敗するにつれ、ステラー博士は、タンパク質の塊は神経変性の原因ではなく、症状なのではないかと考え始めました。
「タンパク質の塊があるのは良いことではありません」とステラー博士は言います。「しかし、人々はあまりにも凝集体に注目しすぎてきました。私たちの研究結果は、それらが病気の原因ではなく、結果であることを示唆しているのです。」
ステラー研究室の以前の研究は、神経変性がタンパク質の塊からではなく、細胞のタンパク質分解装置であるプロテアソームをシナプスへ届けることに失敗することから始まると、長らく示唆してきました。プロテアソームは、細胞本体から神経終末までの長い距離を移動し、そこで定期的に損傷したタンパク質を除去して、ニューロン間の通信を維持しなければなりません。もしプロテアソームが到着しなければ、ゴミが蓄積し、通信は途絶えます。その場合、プラークの除去のみを目的とした治療法では手遅れです。本当の解決策は、渋滞が起こる前に「清掃班」を送り届ける輸送システムを修理することなのです。
2019年の論文で、ステラー博士はその輸送システムを修理するための有望な手がかりを特定しました。それが、プロテアソームを細胞内のモーターに積み込んでシナプスへの旅をさせ、到着時に組み立てる役割を担うアダプターであるタンパク質、PI31です。彼は、PI31がないと輸送が停滞し、タンパク質のゴミが蓄積し、凝集体が形成されることを見出しました。PI31を持たないハエやマウスは神経変性の兆候を示し始め、それ以来、PI31の正常な機能の喪失や低下につながる変異、および関連タンパク質をコードする遺伝子が、多くの神経変性疾患に関与していることが示唆されています。
「PI31をコードする遺伝子の変異は、アルツハイマー病患者に見られます。ALS(筋萎縮性側索硬化症)患者にも見られます。これらの同じ変異を持つ患者は、時にパーキンソン病と診断されることもあります」とステラー博士は言います。「私たちはそれをハエで見て、マウスでノックアウトしました。そこで知りたかったのです。これを治療に使えるだろうか、と。」
新しい治療法に向けて
PI31を増やすことが神経変性を防げるかどうかを検証するため、ステラー博士は、遺伝子FBXO7の変異によって引き起こされる希少な遺伝性疾患に注目しました。これらの変異はヒトで早期発症型のパーキンソン病様症候群を引き起こすため、このモデルは臨床的な関連性を持ちます。同じく重要なことに、FBXO7はPI31と関連しています。FBXO7が失われると、PI31のレベルが低下するのです。
ステラー博士のチームはまずショウジョウバエのモデルから始め、ハエのFBXO7に相当する遺伝子を不活性化すると、予想されるパーキンソン病様の症状と一致して、重度の運動障害を引き起こし、プロテアソームの輸送を妨げることを実証しました。そこにPI31のコピーを追加で加えると、プロテアソームが再びスムーズに動き始め、これらの症状は大部分が回復しました。
次に研究者たちはFBXO7を欠損したマウスに移り、PI31レベルをわずかに増加させるだけでも、神経変性を強力に抑制し、運動機能を維持し、全体的な健康状態を改善することを発見しました。いくつかのケースでは、マウスの寿命がほぼ4倍にまで延びました。PI31はまた、アルツハイマー病の特徴である異常なタウタンパク質も除去しました。
これらの結果は総合的に、PI31の過剰発現がプロテアソームを軌道に乗せ続け、それによってマウスとショウジョウバエにおける神経変性の特徴の多くを防ぐことができることを示しています。「私たちがマウスの様々な欠損を救済できる度合いは驚くべきものです」とステラー博士は言います。
次のステップは、PI31が老化マウスの認知機能を維持できるかどうかをテストし、その後、ヒト向けの治療法の前臨床開発に進むことを目指しています。
最近のプレプリントで、ステラー研究室は、PI31遺伝子に希少な変異を持つヒトが、さまざまな神経変性疾患に苦しんでいることを示すプロジェクトに協力しました。これらの発見は、PI31療法が、これらの疾患に苦しむ少数のヒトにおけるFBXO7またはPI31の欠損によって引き起こされる希少疾患を標的にできる可能性を示しています。いずれは、これらの希少疾患の治療から得られた教訓が、加齢に伴う認知機能の低下を遅らせ、アルツハイマー病のようなより一般的な疾患に取り組むための、より広範な戦略を生み出すだろうとステラー博士は考えています。
「これが私たちのハエやマウスのFBOX7モデルを超えて関連性があることに、私たちは非常に興奮しています」とステラー博士は言います。「この科学は、私たちの発見が将来的には、加齢に伴う認知機能の低下を遅らせることを可能にするかもしれない、ということを示唆しているのです。」
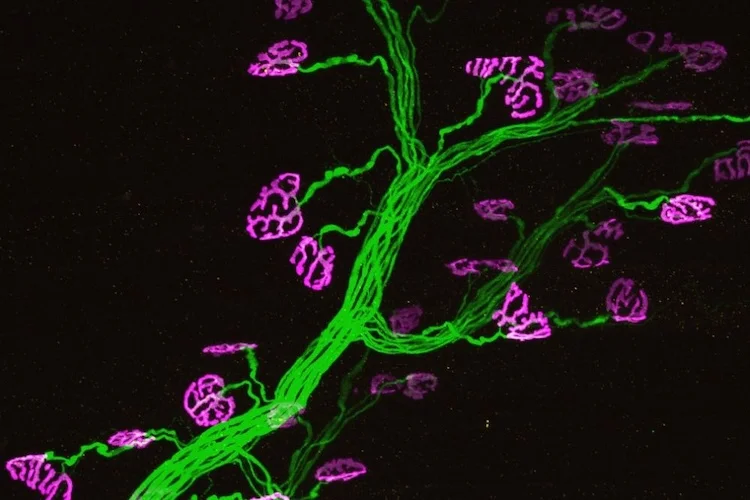
画像:PI31療法により回復した運動ニューロン(緑色の軸索とマゼンタ色の軸索終末を有する)。
(Steller lab)